réservé à la recherche
Triptolide Inhibiteur du Domaine Moyen de Hsp90
N° Cat.S3604
Structure chimique
Poids moléculaire: 360.4
Contrôle qualité
| Cibles apparentées | NF-κB HDAC Antioxidant ROS IκB/IKK Nrf2 AP-1 MALT NOD |
|---|---|
| Autre ADC Cytotoxin Inhibiteurs | SN-38 Luteolin (+)-Bicuculline Rutin Artemisinin BHQ Pinocembrin Harmine hydrochloride Luteoloside Sacituzumab-govitecan |
Culture cellulaire, traitement et concentration de travail
| Lignées cellulaires | Type dessai | Concentration | Temps dincubation | Formulation | Description de lactivité | PMID |
|---|---|---|---|---|---|---|
| HT-29 | Cytotoxicity against | Cytotoxicity against human HT-29 cells, IC50=0.0021μM | 21470864 | |||
| HCT116 | Cytotoxicity against | Cytotoxicity against human HCT116 cells assessed as decrease in cell viability, IC50=0.0047μM | 31121546 | |||
| SKOV3 | Antiproliferative activity against | 72 hrs | Antiproliferative activity against human SKOV3 cells after 72 hrs by SRB assay, IC50=0.006μM | 20833543 | ||
| SKOV3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SKOV3 cells after 72 hrs by sulforhodamine B assay, IC50=0.006μM | 24378709 | ||
| SKOV3 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human SKOV3 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0072μM | 28011223 | ||
| KBM5 | Cytotoxicity against | 72 hrs | Cytotoxicity against imatinib-resistant human KBM5 cells harboring Bcr-Abl T315I mutant after 72 hrs by MTS assay, IC50=0.0083μM | 20149665 | ||
| SKOV3 | Cytotoxicity against | Cytotoxicity against human SKOV3 cells by SRB assay, IC50=0.009μM | 19637874 | |||
| MDA-MB-468 | Cytotoxicity against | Cytotoxicity against human MDA-MB-468 cells by SRB assay, IC50=0.01μM | 19637874 | |||
| HCT116 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HCT116 cells after 72 hrs by MTT assay, IC50=0.01μM | 19637874 | ||
| SKOV3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SKOV3 cells after 72 hrs by MTT assay, IC50=0.01μM | 19637874 | ||
| KBM5 | Cytotoxicity against | 72 hrs | Cytotoxicity against human KBM5 cells harboring wild type Bcr-Abl after 72 hrs by MTS assay, IC50=0.0103μM | 20149665 | ||
| Rh30 | Cytotoxicity against | 72 hrs | Cytotoxicity against human Rh30 cells after 72 hrs by MTT assay, IC50=0.014μM | 19637874 | ||
| A549 | Antagonist activity at | Antagonist activity at human PAR2 expressed in human A549 cells assessed as inhibition of 2f-LIGRLO-NH2-induced NFkappaB activation by luciferase reporter gene assay, IC50=0.014μM | 23895492 | |||
| SGC7901 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SGC7901 cells after 72 hrs by MTT assay, IC50=0.015μM | 19637874 | ||
| MOLT4 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MOLT4 cells after 72 hrs by MTT assay, IC50=0.017μM | 19637874 | ||
| A549 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human A549 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0175μM | 28011223 | ||
| SMMC7721 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SMMC7721 cells after 72 hrs by MTT assay, IC50=0.018μM | 19637874 | ||
| PC3 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human PC3 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0183μM | 28011223 | ||
| MCF7 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50=0.019μM | 19637874 | ||
| A549 | Cytotoxicity against | Cytotoxicity against human A549 cells, IC50=0.019μM | 21470864 | |||
| PC3 | Cytotoxicity against | Cytotoxicity against human PC3 cells by SRB assay, IC50=0.02μM | 19637874 | |||
| Bel7402 | Cytotoxicity against | 72 hrs | Cytotoxicity against human Bel7402 cells after 72 hrs by MTT assay, IC50=0.02μM | 19637874 | ||
| PC3 | Antiproliferative activity against | 72 hrs | Antiproliferative activity against human PC3 cells after 72 hrs by SRB assay, IC50=0.02μM | 20833543 | ||
| PC3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by sulforhodamine B assay, IC50=0.02μM | 24378709 | ||
| PC3 | Cytotoxicity against | Cytotoxicity against human PC3 cells assessed as inhibition of cell proliferation by sulforhodamine B assay, IC50=0.02μM | 25467158 | |||
| 786-O | Cytotoxicity against | 72 hrs | Cytotoxicity against human 786-O cells after 72 hrs by MTT assay, IC50=0.022μM | 19637874 | ||
| A549 | Antagonist activity at | Antagonist activity at human PAR2 expressed in human A549 cells coexpressing TACR1 assessed as inhibition of substance P-induced IL-8 production by ELISA, IC50=0.023μM | 23895492 | |||
| MDA-MB-231 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MDA-MB-231 cells after 72 hrs by MTT assay, IC50=0.024μM | 19637874 | ||
| DU145 | Cytotoxicity against | 72 hrs | Cytotoxicity against human DU145 cells after 72 hrs by MTT assay, IC50=0.024μM | 19637874 | ||
| HO8910 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HO8910 cells after 72 hrs by MTT assay, IC50=0.028μM | 19637874 | ||
| HCT15 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HCT15 cells after 72 hrs by MTT assay, IC50=0.029μM | 19637874 | ||
| A549 | Growth inhibition of human | Growth inhibition of human A549 cells, IC50=0.03μM | 28814374 | |||
| 32D | Cytotoxicity against | 72 hrs | Cytotoxicity against mouse 32D cells harboring wild type Bcr-Abl after 72 hrs by MTS assay, IC50=0.032μM | 20149665 | ||
| U251 | Cytotoxicity against | Cytotoxicity against human U251 cells assessed as inhibition of cell proliferation by sulforhodamine B assay, IC50=0.033μM | 25467158 | |||
| 32D | Cytotoxicity against | 72 hrs | Cytotoxicity against imatinib-resistant mouse 32D cells harboring Bcr-Abl T315I mutant after 72 hrs by MTS assay, IC50=0.034μM | 20149665 | ||
| PC3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by MTT assay, IC50=0.043μM | 19637874 | ||
| KB | Cytotoxicity against | 72 hrs | Cytotoxicity against human KB cells after 72 hrs by MTT assay, IC50=0.043μM | 19637874 | ||
| HepG2 | Cytotoxicity against | 48 hrs | Cytotoxicity against human HepG2 cells after 48 hrs by XTT assay, IC50=0.0433μM | 30613335 | ||
| HeLa | Cytotoxicity against | 72 hrs | Cytotoxicity against human HeLa cells after 72 hrs by MTT assay, IC50=0.047μM | 19637874 | ||
| U251 | Cytotoxicity against | 72 hrs | Cytotoxicity against human U251 cells after 72 hrs by MTT assay, IC50=0.049μM | 19637874 | ||
| K562 | Cytotoxicity against | 72 hrs | Cytotoxicity against human K562 cells after 72 hrs by MTT assay, IC50=0.05μM | 19637874 | ||
| NIH/3T3 | Cytotoxicity against | Cytotoxicity against mouse NIH/3T3 cells assessed as decrease in cell viability, IC50=0.05μM | 31121546 | |||
| SW1116 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SW1116 cells after 72 hrs by MTT assay, IC50=0.052μM | 19637874 | ||
| A549 | Cytotoxicity against | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50=0.059μM | 19637874 | ||
| HeLa | Cytotoxicity against | Cytotoxicity against human HeLa cells assessed as decrease in cell viability, IC50=0.087μM | 31121546 | |||
| Jurkat | Cytotoxicity against | Cytotoxicity against human Jurkat cells assessed as decrease in cell viability, IC50=0.14μM | 31121546 | |||
| MKN28 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MKN28 cells after 72 hrs by MTT assay, IC50=0.2μM | 19637874 | ||
| MDCK | Cytotoxicity against | Cytotoxicity against MDCK cells assessed as decrease in cell viability, IC50=1.2μM | 31121546 | |||
| Pkd1-/- | Induction of | Induction of cell growth arrest in mouse Pkd1-/- cells in presence of calcium | 17360534 | |||
| Pkd1-/- | Increase in | 100 nM | 96 hrs | Increase in p21CIP/WAF expression in mouse Pkd1-/- cells at 100 nM after 96 hrs by Western blot analysis | 17360534 | |
| Pkd1+/- | Increase in | 100 nM | Increase in PC2 dependent calcium release in mouse Pkd1+/- cells at 100 nM | 17360534 | ||
| Pkd1-/- | Increase in | 100 nM | Increase in PC2 dependent calcium release in mouse Pkd1-/- cells at 100 nM | 17360534 | ||
| Pkd1-/- | Increase in | 50 uM | Increase in calcium release in mouse Pkd1-/- cells at 50 uM in presence of RyR antagonist dantrolene | 17360534 | ||
| Pkd2+/- | Growth inhibition of PC2 expressing mouse | 100 nM | 24 hrs | Growth inhibition of PC2 expressing mouse Pkd2+/- cells as cell death at 100 nM after 24 hrs | 17360534 | |
| KBM5 | Cytotoxicity against | 0.001 to 17 uM | 72 hrs | Cytotoxicity against human KBM5 cells harboring wild type Bcr-Abl at 0.001 to 17 uM after 72 hrs by MTS assay | 20149665 | |
| KBM5 | Cytotoxicity against | 0.001 to 17 uM | 72 hrs | Cytotoxicity against imatinib-resistant human KBM5 cells harboring Bcr-Abl T315I mutant at 0.001 to 17 uM after 72 hrs by MTS assay | 20149665 | |
| LNCAP | Antagonist activity at | 5 uM | 24 hrs | Antagonist activity at AR in human LNCAP cells assessed as suppression of DHT-induced receptor transcriptional activity at 5 uM after 24 hrs by dual luciferase reporter gene assay | 27994731 | |
| LNCAP | Antagonist activity at | 500 nM | 24 hrs | Antagonist activity at AR in human LNCAP cells assessed as suppression of DHT-induced receptor transcriptional activity at 500 nM after 24 hrs by dual luciferase reporter gene assay | 27994731 | |
| HepG2 | Antitumor activity against | 0.2 mg/kg | 15 days | Antitumor activity against human HepG2 cells xenografted in Balb/c nude mouse assessed as reduction in tumor growth at 0.2 mg/kg, ip administered once daily for 15 days | 30613335 | |
| Cliquez pour voir plus de données expérimentales sur les lignées cellulaires | ||||||
Informations chimiques, stockage et stabilité
| Poids moléculaire | 360.4 | Formule | C20H24O6 |
Stockage (À partir de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 38748-32-2 | Télécharger le SDF | Stockage des solutions mères |
|
|
Solubilité
|
In vitro |
DMSO
: 72 mg/mL
(199.77 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Entrez les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Entrez la formulation in vivo (Ceci nest que le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuiteμL PEG300, mélanger et clarifier, ajouter ensuiteμL Tween 80, mélanger et clarifier, ajouter ensuite μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, ajouter ensuite μL Huile de maïs, mélanger et clarifier.
Remarque : 1. Assurez-vous que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue lors de lajout précédent est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
NF-κB
HSF1
MDM2
|
|---|---|
| In vitro |
Le Triptolide est un tri-époxyde diterpénique doté de puissantes propriétés immunosuppressives et anti-inflammatoires. Ce composé est montré pour inhiber l'expression de l'IL-2 dans les lymphocytes T activés au niveau du facteur purine-box/nucléaire et de l'activation de la transcription médiée par NF-κB. Il inhibe la prolifération et la formation de colonies de cellules tumorales à des concentrations extrêmement faibles (2–10 ng/mL). Ce produit chimique a une activité inhibitrice sur les cellules de cancer du sein, de l'estomac et de la lignée cellulaire de leucémie HL-60. Il induit l'apoptose dans les cellules tumorales en bloquant l'activation de NF-κB et en sensibilisant les cellules tumorales à la mort cellulaire programmée induite par le TNF-α.
|
| In vivo |
Le Triptolide agit en synergie avec la cyclosporine A pour favoriser la survie des greffons dans des modèles animaux et pour supprimer la maladie du greffon contre l'hôte dans les greffes allogéniques de moelle osseuse. De plus, il induit l'apoptose dans les cellules tumorales et potentialise l'induction de l'apoptose par le facteur de nécrose tumorale (TNF-α), en partie par la suppression de l'induction de c-IAP2 et c-IAP1. Ce traitement composé pendant 2 à 3 semaines inhibe la croissance de xénogreffes formées par quatre lignées cellulaires tumorales différentes (mélanome B16, cancer du sein MDA-435, cancer de la vessie TSU et carcinome gastrique MGC80-3), indiquant que le TPL a un large spectre d'activité contre les tumeurs qui contiennent des formes sauvages et mutantes de p53. De plus, il inhibe la métastase expérimentale des cellules B16F10 vers les poumons et les rates de souris. Il a des activités in vitro et in vivo contre des modèles murins de maladie rénale polykystique. DL50 : Souris 0,83 mg/kg (i.v.).
|
Références |
|
Applications
| Méthodes | Biomarqueurs | Images | PMID |
|---|---|---|---|
| Western blot | c-Jun MDM2 p-AKT / AKT / p-Foxo3a / Foxo3a / p53 p-PI3K / PI3K / p85 / p110 |
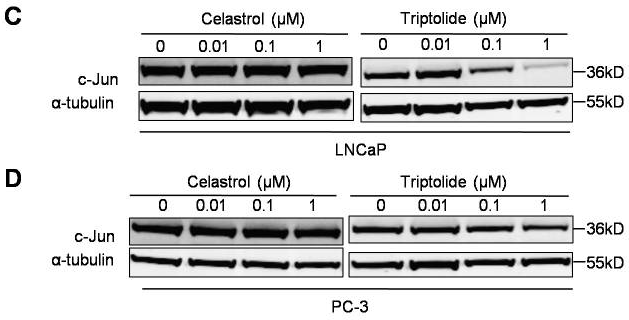
|
22666381 |
| Growth inhibition assay | Cell viability |
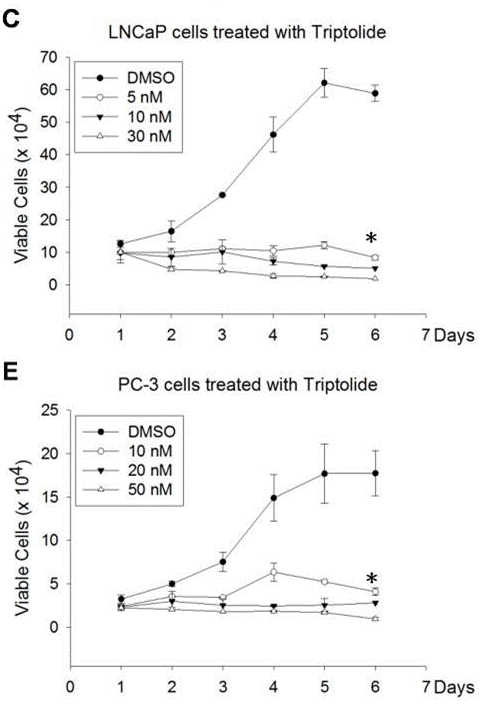
|
22666381 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Non destinés à lusage humain. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.