Bcl-2 (B-cell lymphoma 2) is the founding pro-survival member of the Bcl-2 protein family, exerting its pro-survival function in response to a broad range of apoptotic stimuli through the inhibition of the mitochondrial outer membrane permeabilization (MOMP) process and the release of mitochondrial cytochrome c. [show the full text]
Bcl-2 억제제 (Bcl-2 Inhibitors)
기타 Apoptosis 억제제
| Cat.No. | 제품명 | 정보 | 제품 사용 인용 | 제품 검증 |
|---|---|---|---|---|
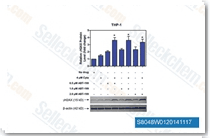
| S8048 | ABT-199 (Venetoclax) | Venetoclax (ABT-199, GDC-0199)는 세포가 없는 분석에서 Ki가 <0.01 nM인 Bcl-2 선택적 억제제로, Bcl-xL 및 Bcl-w에 대해 4800배 이상 더 선택적이며, Mcl-1에는 활성이 없습니다. Venetoclax는 삼중 음성 유방암 MDA-MB-231 세포에서 세포 성장 억제, apoptosis, 세포 주기 정지 및 autophagy를 유도하는 것으로 보고되었습니다. 3상. |
|
|
| S1001 | Navitoclax (ABT-263) | 무세포 분석에서 Ki가 ", "0.5 nM, ", "1 nM 및 ", "1 nM인 Bcl-xL, Bcl-2 및 Bcl-w의 강력한 억제제인 Navitoclax (ABT-263)는 Mcl-1 및 A1에 더 약하게 결합합니다. 2상. |
|

|
| S8383 | S63845 | S63845는 Kd 값 0.19 nM을 가진 새로운 선택적 MCL-1 억제제로, 다른 Bcl-2 계열인 Bcl-2 또는 BCL-XL에는 식별 가능한 결합을 보이지 않습니다. |
|
|
| S1002 | ABT-737 | ABT-737은 BH3 모방 억제제로, Bcl-xL, Bcl-2 및 Bcl-w에 대해 각각 78.7 nM, 30.3 nM 및 197.8 nM의 EC50을 세포 없는 분석에서 나타냅니다. Mcl-1, Bcl-B 또는 Bfl-1에 대해서는 억제가 관찰되지 않았습니다. ABT-737은 미토콘드리아 경로 Apoptosis 및 Mitophagy를 유도합니다. 2상 연구 중입니다. |
|

|
| S7747 | Ro-3306 | RO-3306은 Ki가 20 nM인 ATP 경쟁적이고 선택적인 CDK1 억제제로, 다양한 인체 키나제 패널에 대해 15배 이상의 선택성을 보입니다. RO-3306은 p53 매개 Bax 활성화 및 미토콘드리아 아폽토시스를 증강합니다. |
|

|
| S1057 | Obatoclax Mesylate (GX15-070) | Obatoclax Mesylate (GX15-070)는 세포 유리 분석에서 Ki가 0.22 μM인 Bcl-2 길항제이며, MCL-1 매개 Apoptosis 저항성을 극복하는 데 도움을 줄 수 있습니다. |
|

|
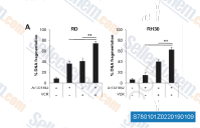
| S7801 | A-1331852 | A-1331852는 BCL-XL에 대해 0.01 nM 미만, Bcl-2, Bcl-W, MCL-1에 대해 각각 6 nM, 4 nM, 142 nM의 Ki 값을 갖는 강력하고 선택적인 BCL-XL 억제제입니다. 암, 면역 및 자가면역 질환 치료에 유용할 수 있습니다. |
|
|
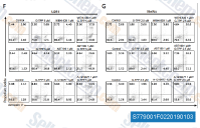
| S7790 | A-1210477 | A-1210477은 Ki 및 IC50이 각각 0.454 nM 및 26.2 nM인 강력하고 선택적인 MCL-1 억제제로, 다른 Bcl-2 계열 구성원에 비해 >100배의 선택성을 보입니다. |
|
|
| S1121 | TW-37 | TW-37은 재조합 Bcl-2, Bcl-xL 및 Mcl-1에 대한 새로운 비펩타이드 억제제로, 무세포 분석에서 각각 0.29 μM, 1.11 μM 및 0.26 μM의 Ki를 갖습니다. |
|

|
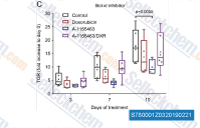
| S7800 | A-1155463 Dihydrochloride | 고도로 강력하고 선택적인 BCL-XL 억제제인 A-1155463 Dihydrochloride는 BCL-XL에 대해 피코몰 결합 친화도를 나타내며, BCL-2 및 관련 단백질 BCL-W(Ki=19 nM) 및 MCL-1(Ki>440 nM)에는 >1000배 약한 결합을 나타냅니다. |
|
|
Bcl-2 (B-cell lymphoma 2) is encoded by the Bcl-2 gene and is the first identified member of a large family of apoptosis regulatory proteins (Bcl-2 family) that derives its name from the B-cell lymphoma 2, as it is the second member of a variety of proteins initially described in the t(14;18) chromosomal translocation in human follicular B-cell lymphomas. Bcl-2 contains four Bcl-2 homology domains (BH1-BH4) that mediate the formation of homodimer and heterodimer with relative proteins such as Bax, Bad, Bak and Bcl-xL, and a trans-membrane (TM) domain that mediates insertion into the outer membrane of the mitochondria and the endoplasmic reticulum. Bcl-2 proteins are generally integrated within the outer mitochondrial membrane (OMM), and may also be in the cytosol or ER membrane. The Bcl-2 and other antiapoptotic members of the Bcl-2 family preserve the outer mitochondrial membrane (OMM) integrity, thus inhibiting the mitochondrial signaling pathway of apoptosis, by complex interactions with the proapoptotic Bcl-2 proteins such as Bax, Bak, Bim, Puma and tBid. [1][2]
Bcl-2 suppresses apoptosis in response to a broad range of stress stimuli, including those frequently encountered during tumor development, such as oncogene activation, DNA damage, hypoxia (oxygen deprivation), loss of appropriate growth signals and anoikis (loss of cell attachment). In healthy cells, Bax and Bak are kept in check by the pro-survival Bcl-2 family members and the binding of BH3-only proteins unleashes Bax/Bak. Bcl-2 is also critical for the survival of renal epithelial stem cells during embryogenesis, melanocyte progenitors and mature B and T lymphocytes. Bcl-2 over-expression accelerates Eu-myc-induced lymphomagenesis, but loss of endogenous Bcl-2 does not prevent or delay Eu-myc-induced B lymphoma development. Bcl-2 proteins also constitutively binds to Beclin-1, and its dissociation through post-translational modification of Beclin-1 and/or Bcl-2 proteins such as phosphorylation by JNK1, or direct competition for the Bcl-2 BC groove by another BH3-only protein such as Bad, may be sufficient to induce autophagy, leading to the suggestion that autophagy and apoptosis are mechanistically linked. Single-site phosphorylation at Serine 70 (S70) is required for the antiapoptotic function of Bcl-2, and multisite phosphorylation at Threonine 69, S70, and S87 has been reported to inactivate Bcl-2. Phosphorylation of Bcl-2 has been shown to enhance activity to allow response to extracellular growth-factor-mediated signals. [1][2][3]
In addition, Bcl-2 is over-expressed in human follicular centre B-cell lymphoma; high levels of Bcl-2 are also detected in significant numbers of chronic lymphocytic leukaemia (CLL), DLBCL and mantle cell lymphoma, as well as in certain solid tumours(brain, breast and lung). The upregulation of Bcl-2 in CLL and other cancers has been attributed to the hypo-methylation of the Bcl-2 promoter or, possibly more importantly due to hemizygous or homozygous loss of the micro RNAs (miRs) 15a and 16-1 that negatively regulate Bcl-2. The dysregulated Bcl-2 proteins in cancer can lead to increased survival of abnormal cells, which are thought to be involved in resistance to conventional cancer treatment. Mice that constitutively express both Myc and Bcl-2 transgenes develop lymphoblastic leukaemia with high incidence, while shut-down of the inducible Bcl-2 transgene in lymphoma-burdened bi-transgenic mice results in tumor regression and significantly prolonged animal survival in many cases, indicating that inactivation of Bcl-2 constitutes a promising new approach to cancer therapy. Small molecule mimetics of BH3-only proteins that can directly target pro-survival Bcl-2 family members are being developed as a novel therapeutic approach. ABT-737 and the closely related orally bioavailable ABT-263, belong to the BH3 mimetic small molecule inhibitors, targeting Bcl-2 and Bcl-2-related proteins such as Bcl-xL and Bcl-w, therefore promoting tumor regression in murine xeno-transplanation models of certain human lymphomas or small cell lung carcinomas and in primary patient-derived follicular lymphoma cells. [1][4]
제품은 연구용으로만 사용됩니다. 인체에는 사용하지 마십시오. 환자에게 판매하지 않습니다.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.