ERK inhibitors are a class of targeted therapeutic agents that specifically modulate the extracellular signal-regulated kinase (ERK) pathway, a core component of the mitogen-activated protein kinase (MAPK) signaling cascade. The ERK pathway is evolutionarily conserved and plays a pivotal role in transducing extracellular signals (e.g., growth factors, cytokines) into intracellular responses, regulating critical cellular processes such as proliferation, differentiation, survival, and migration. Dysregulation of the ERK pathway, often driven by mutations in upstream components, is a hallmark of numerous human diseases, particularly cancers, making ERK inhibitors a central focus of translational research. This article explores the scientific underpinnings of ERK inhibitors, including their mechanism of action, interactions with related signaling molecules such as MEK and AKT, and research progress exemplified by SCH series inhibitors.
ERK 억제제/활성제 (ERK Inhibitors/Activators)
| Cat.No. | 제품명 | 정보 | 제품 사용 인용 | 제품 검증 |
|---|---|---|---|---|
| S7554 | Ravoxertinib (GDC-0994) | Ravoxertinib (GDC-0994)는 각각 1.1 nM 및 0.3 nM의 IC50 값을 갖는 강력하고 경구 투여 가능하며 고도로 선택적인 ERK1/2 억제제입니다. 이 화합물은 현재 1상 임상시험 중입니다. |
|

|
| S7854 | Ulixertinib (BVD-523) | Ulixertinib (BVD-523, VRT752271)은 ERK2에 대해 IC50이 <0.3 nM인 강력하고 가역적인 ERK1/ERK2 억제제입니다. 이 화합물은 1상에 있습니다. |
|

|
| S7101 | SCH772984 | SCH772984는 무세포 분석에서 각각 4nM 및 1nM의 IC50 값을 갖는 ERK1/2의 새로운, 특이적 억제제이며, RAS- 또는 BRAF-돌연변이 암세포에서 강력한 효능을 보인다. |
|

|
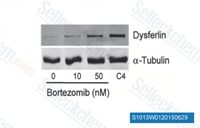
| S1013 | Bortezomib | Bortezomib은 Ki가 0.6 nM인 강력한 20S proteasome 억제제입니다. 이는 정상 세포보다 종양 세포에 대해 유리한 선택성을 나타냅니다. 이 화합물은 NF-κB를 억제하고 ERK 인산화를 유도하여 Cathepsin B를 억제하고 난소암 및 기타 고형암에서 autophagy의 촉매 과정을 억제합니다. |
|
|
| S9698 | Ezatiostat | 글루타티온의 트리펩티드 유사체인 Ezatiostat는 Glutathione S-transferase P1-1 (GSTP1-1)의 펩티도모방 억제제입니다. 이 화합물은 c-Jun NH2 terminal kinase (JNK1)와 ERK1/ERK2를 활성화하고 Apoptosis를 유도합니다. | ||
| S1396 | Resveratrol (trans-Resveratrol) | Resveratrol은 시클로옥시게나제(예: COX, IC50=1.1 μM), 리폭시게나제(LOX, IC50=2.7 μM), 키나제, 시르투인 및 기타 단백질을 포함한 광범위한 표적을 가지고 있습니다. 항암, 항염증, 혈당 강하 및 기타 유익한 심혈관 효과를 가집니다. Resveratrol은 mitophagy/autophagy 및 자가포식 의존성 apoptosis를 유도합니다. |
|
|
| S8534 | LY3214996 (Temuterkib) | Temuterkib (LY3214996)는 생화학적 분석에서 두 효소에 대해 IC50이 5nM인 선택적이고 새로운 ERK1/2 억제제입니다. BRAF 및 RAS 돌연변이 암 세포주에서 세포 phospho-RSK1을 강력하게 억제합니다. |
|
|
| S7524 | FR 180204 | FR 180204는 ATP 경쟁적인 선택적 ERK 억제제로, ERK1 및 ERK2에 대한 Ki는 각각 0.31 μM 및 0.14 μM입니다. 관련 키나아제 p38α에 대해서는 30배 덜 강력하며, 30 μM 미만에서는 어떠한 키나아제(MEK1, MKK4, IKKα, PKCα, Src, Syc 및 PDGFα)도 억제하지 못했습니다. |
|

|
| S7709 | VX-11e | VX-11e (VTX-11e, Vertex-11e)는 강력하고 선택적이며 경구 생체이용률이 높은 ERK 억제제로, IC50는 17 nM (ERK1) 및 15 nM (ERK2)이며, 테스트된 다른 키나아제에 비해 200배 이상 선택적입니다. |
|
|
| S7525 | XMD8-92 | XMD8-92는 big map kinase (BMK1, ERK5) 및 bromodomain-containing proteins (BRDs, BET)의 강력하고 선택적인 이중 억제제로, ERK5 및 BRD4(1)에 대한 Kd는 각각 80 nM 및 170 nM입니다. |
|
|
Mechanism of Action: Targeting ERK in the MAPK Pathway
The ERK pathway (also known as the Raf-MEK-ERK pathway) operates through a sequential phosphorylation cascade: upstream signals activate Raf kinases, which phosphorylate and activate MEK (mitogen-activated protein kinase kinase) 1/2, and activated MEK then phosphorylates ERK1/2. Phosphorylated ERK (p-ERK) translocates to the nucleus, where it phosphorylates a variety of transcription factors (e.g., ELK1, c-Myc) and regulatory proteins, ultimately modulating gene expression. ERK inhibitors disrupt this cascade by targeting ERK1/2, the terminal kinase in the pathway, distinguishing them from upstream inhibitors targeting Raf or MEK. This unique targeting position endows ERK inhibitors with distinct advantages, particularly in overcoming resistance to upstream inhibitors.
Direct ERK Inhibition: Competing with ATP and Blocking Phosphorylation
Most clinically advanced ERK inhibitors act as ATP-competitive inhibitors, binding to the ATP-binding pocket of ERK1/2 to prevent MEK-mediated phosphorylation and subsequent activation. Unlike MEK inhibitors, which block ERK activation indirectly, direct ERK inhibitors can also suppress the activity of constitutively active ERK mutants (e.g., ERK2) that emerge as resistance mechanisms to MEK inhibition. Additionally, some ERK inhibitors exhibit "dual-action" properties: in addition to blocking kinase activity, they promote the degradation of activated ERK through ubiquitin-proteasome pathways. Preclinical studies demonstrate that this dual mechanism enhances inhibitory efficacy, as it not only prevents ERK activation but also reduces the pool of active ERK molecules, minimizing the risk of pathway reactivation.
1.2 Indirect ERK Modulation: Targeting Scaffold Proteins and Substrates
A growing area of research focuses on indirect ERK inhibitors that target proteins involved in ERK localization, scaffolding, or substrate interaction, rather than the kinase domain itself. For example, scaffold proteins such as KSR1 (kinase suppressor of Ras) facilitate the assembly of the Raf-MEK-ERK complex, enhancing pathway activation. Inhibitors targeting KSR1 disrupt complex formation, reducing ERK phosphorylation without directly binding to ERK. Another strategy involves targeting ERK-substrate interactions: small molecules that bind to the docking domain of ERK can block its interaction with specific transcription factors, allowing for selective modulation of ERK-dependent gene expression. This indirect approach offers potential for reduced off-target effects, as it targets context-specific ERK functions rather than global kinase activity.
Interactions with MEK and AKT: Synergy and Resistance Modulation
The ERK pathway does not operate in isolation; it crosstalks extensively with other signaling cascades, most notably the PI3K-AKT pathway. Additionally, as MEK is the immediate upstream activator of ERK, the interaction between ERK and MEK inhibitors is critical for therapeutic efficacy. Understanding these interactions is essential for optimizing ERK inhibitor-based therapies and overcoming resistance.
ERK-MEK Feedback Loops and Combination Therapy
A key challenge in targeting the ERK pathway is the presence of negative feedback loops between ERK and MEK. For instance, activated ERK phosphorylates and inhibits Raf, creating a negative feedback that restrains pathway activity. MEK inhibitors disrupt this feedback, leading to paradoxical activation of Raf and subsequent ERK reactivation—a major mechanism of resistance. ERK inhibitors, by contrast, directly suppress ERK activity without disrupting the feedback loop, making them effective in overcoming MEK inhibitor resistance. Preclinical and clinical studies show that combining ERK and MEK inhibitors synergistically suppresses pathway activation, as MEK inhibitors block ERK activation upstream and ERK inhibitors prevent residual or reactivated ERK activity. This combination strategy has shown promise in cancers with BRAF or RAS mutations, such as melanoma and colorectal cancer.
Crosstalk with the AKT Pathway: Implications for Therapy
The ERK and AKT pathways exhibit extensive crosstalk, with mutual activation or inhibition influencing therapeutic response. For example, inhibition of ERK can lead to upregulation of the PI3K-AKT pathway through increased expression of growth factor receptors (e.g., EGFR) or activation of downstream adaptor proteins. This compensatory AKT activation reduces the efficacy of ERK inhibitors, particularly in cancers with coexisting mutations in PI3K pathway components. Conversely, AKT inhibition can enhance ERK inhibitor efficacy by blocking this compensatory pathway. Preclinical models of pancreatic cancer and non-small cell lung cancer (NSCLC) demonstrate that combining ERK inhibitors with AKT inhibitors or PI3K inhibitors significantly reduces tumor growth compared to single-agent therapy. Additionally, biomarkers of AKT pathway activation (e.g., phosphorylated AKT, PTEN loss) may predict response to combination therapy, highlighting the need for personalized approaches.
Common ERK Inhibitors: Preclinical Research and Clinical Progress
A variety of ERK inhibitors have been developed and advanced through preclinical and clinical research, serving as important tools for exploring ERK pathway biology and potential therapeutic agents for pathway-dysregulated diseases. Among these, compounds such as SCH772984, ulixertinib (SCH900353), and KO-947 have been widely studied, providing critical insights into the mechanism of action, efficacy, and resistance of ERK inhibitors, and laying the foundation for the development of subsequent inhibitors.
SCH772984: A Classic Tool Compound for Mechanistic Research
SCH772984 is a potent, selective ATP-competitive inhibitor of ERK1/2 (IC values of 4 nM and 1 nM for ERK1 and ERK2, respectively) that has become a widely used tool compound in preclinical research. Studies using SCH772984 have elucidated key aspects of ERK function, including its role in cell cycle progression, apoptosis, and epithelial-mesenchymal transition (EMT). For example, SCH772984 treatment induces G1 cell cycle arrest and apoptosis in BRAF-mutant melanoma cells, while reducing EMT and metastasis in NSCLC models. Additionally, SCH772984 has been used to identify resistance mechanisms, such as mutations in the ERK ATP-binding pocket (e.g., ERK2) and upregulation of the IGF-1R-AKT pathway. These findings have guided the design of next-generation ERK inhibitors with improved resistance profiles.
Ulixertinib (SCH900353): From Preclinical Efficacy to Clinical Trials
Ulixertinib (SCH900353) is an orally bioavailable ERK inhibitor derived from SCH772984, optimized for clinical use with enhanced pharmacokinetic properties and reduced off-target activity. Preclinical studies show that ulixertinib effectively suppresses ERK phosphorylation in a variety of cancer models, including those resistant to BRAF or MEK inhibitors. Early-phase clinical trials (Phase I/II) in patients with advanced solid tumors (e.g., melanoma, colorectal cancer, NSCLC) harboring RAS or BRAF mutations demonstrated promising efficacy: objective response rates (ORR) of 15-20% and disease control rates (DCR) of 50-60% in heavily pretreated patients. Common adverse events included fatigue, diarrhea, and rash, which were manageable with dose adjustments. However, acquired resistance remains a challenge, with studies identifying mutations in ERK2 and activation of alternative signaling pathways (e.g., MAPKAPK2) as key mechanisms. Ongoing trials are exploring ulixertinib in combination with MEK inhibitors, AKT inhibitors, and immunotherapies to improve outcomes.
KO-947: A Promising ERK Inhibitor with Unique Pharmacological Profiles
KO-947 is another notable ERK inhibitor that has garnered attention in preclinical and early clinical research due to its high selectivity and favorable pharmacokinetic properties. As a potent ATP-competitive inhibitor of ERK1/2, KO-947 exhibits low nanomolar IC values (IC50 for ERK1: ~2 nM; ERK2: ~1 nM), comparable to other leading ERK inhibitors, but with minimal cross-reactivity with other kinase families, which contributes to its improved safety profile in preclinical models. A key feature of KO-947 is its ability to penetrate the blood-brain barrier (BBB), a property rarely observed in first-generation ERK inhibitors. This makes it a particularly promising candidate for the treatment of central nervous system (CNS) metastases, which are common in cancers such as melanoma and NSCLC and often refractory to conventional therapies.
In conclusion, ERK inhibitors have emerged as valuable tools in biomedical research and promising therapeutic agents for diseases driven by ERK pathway dysregulation. Their unique mechanism of action, particularly in overcoming resistance to upstream inhibitors, and synergistic potential with MEK or AKT inhibitors highlight their clinical relevance. Widely studied compounds such as SCH772984, ulixertinib, and KO-947 have been instrumental in advancing our understanding of ERK biology and inhibitor development, with ulixertinib and KO-947 paving the way for clinical application—especially KO-947's potential in treating CNS metastases. Future research will focus on optimizing combination strategies, identifying predictive biomarkers, and developing next-generation inhibitors to overcome resistance, further unlocking the therapeutic potential of ERK targeting.
제품은 연구용으로만 사용됩니다. 인체에는 사용하지 마십시오. 환자에게 판매하지 않습니다.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.